High-Energy Broadly Tunable Picosecond OPG PGx01
High Energy Broadly Tunable Picosecond OPG
Travelling Wave Optical Parametric Generators (TWOPG) are an excellent choice for researchers who need an ultra‑fast tunable coherent light source from UV to mid IR.

| Model | PG401 | PG401-SH | PG401-DUV |
|---|---|---|---|
| OPA specifications 1) | |||
| Tuning range | |||
| DUV | – | – | 193 – 209.95 nm |
| SH | – | 210 – 340 nm, 370 – 419 nm | – |
| Signal | 410 – 680 nm | – | – |
| Idler | 740 – 2300 nm | – | – |
| Output pulse energy 2) | > 1000 µJ at 450 nm | > 100 µJ at 300 nm | > 50 µJ at 200 nm |
| Linewidth | < 6 cm‑1 | < 9 cm‑1 | < 9 cm‑1 |
| Max pulse repetition rate | 50 Hz | 50 Hz | 50 Hz |
| Scanning step | |||
| Signal | 0.1 nm | – | – |
| Idler | 1 nm | – | – |
| Typical beam size 3) | ~ 4 mm | ~ 3 mm | ~ 3 mm |
| Beam divergence 4) | < 2 mrad | < 2 mrad | < 2 mrad |
| Beam polarization | |||
| Signal | horizontal | – | – |
| Idler | horizontal | – | – |
| OPG | – | vertical | vertical |
| Typical pulse duration | ~20 ps | ~20 ps | ~20 ps |
| Pump laser requirements | |||
| Pump energy | |||
| at 355 nm | 10 mJ | 10 mJ | 10 mJ |
| at 1064 nm | – | – | 2 mJ |
| Recommended pump source 5) | PL2231-50-TH, PL2251A-TH | PL2231-50-TH, PL2251A-TH | PL2231-50-TH, PL2251A-TH |
| Beam divergence | < 0.5 mrad | < 0.5 mrad | < 0.5 mrad |
| Beam profile | homogeneous, without hot spots, Gaussian fit >90 % | homogeneous, without hot spots, Gaussian fit >90 % | homogeneous, without hot spots, Gaussian fit >90 % |
| Pulse duration 6) | 29 ± 5 ps | 29 ± 5 ps | 29 ± 5 ps |
| Physical characteristics | |||
| Size (W x L x H) | 456 × 633 × 244 mm | 456 × 1031 × 249 ± 3 mm | 456 × 1031 × 249 ± 3 mm |
| Operating requirements | |||
| Room temperature | 15 – 30 °C | 15 – 30 °C | 15 – 30 °C |
| Power requirements | 100 – 240 V AC single phase, 47 – 63 Hz | 100 – 240 V AC single phase, 47 – 63 Hz | 100 – 240 V AC single phase, 47 – 63 Hz |
| Power consumption | < 100 W | < 100 W | < 100 W |
| Model | PG401 | PG401-SH | PG401-DUV |
|---|
- Due to continuous improvement, all specifications are subject to change without notice. Parameters marked typical are not specifications. They are indications of typical performance and will vary with each unit we manufacture. Unless stated otherwise, all specifications are measured at 450 nm for PG401 units, and 300 nm for PG401SH units and for basic system without options.
- See tuning curves for typical pulse energies at other wavelengths. Higher energies are available, please contact Ekspla for more details.
- Beam diameter is measured at the 1/e² level.
- Full angle measured at the FWHM point.
- If a pump laser other than PL2250 or PL2230 is used, measured beam profile data should be presented when ordering.
- Should be specified if non-EKSPLA pump laser is used.
Note: Laser must be connected to the mains electricity all the time. If there will be no mains electricity for longer that 1 hour then laser (system) needs warm up for a few hours before switching on.

Ordering information of PG4xx OPG.

Typical PG401 model tuning curve.
Pump energy: 10 mJ at 355 nm.

Typical PG401-SH model tuning curve.
Pump energy: 10 mJ at 355 nm.

Typical PG401-DUV model tuning curve.
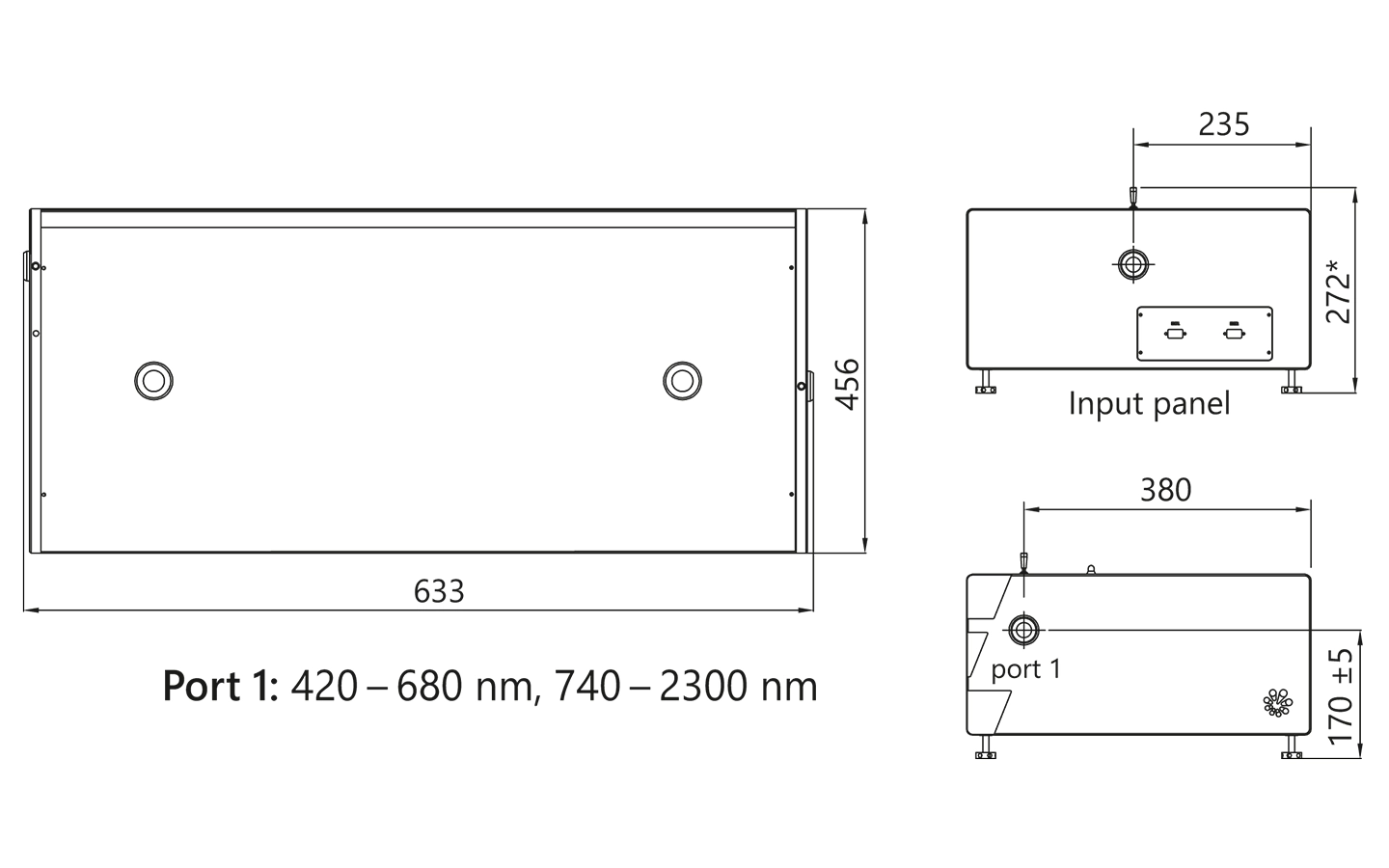
PG401 external dimensions.
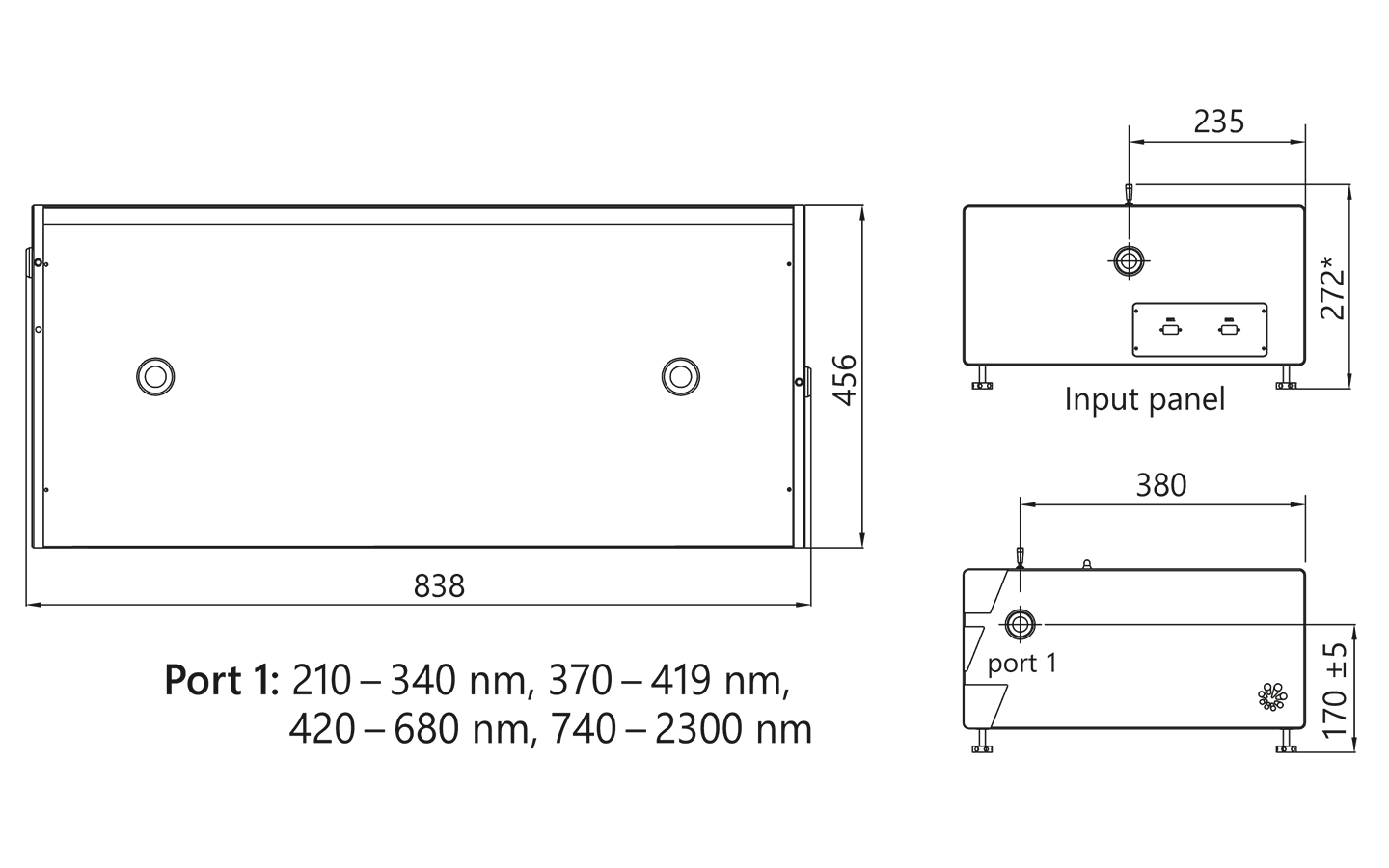
PG401-SH external dimensions.

PG401-SH/DUV external dimensions.
Recommended optical layouts
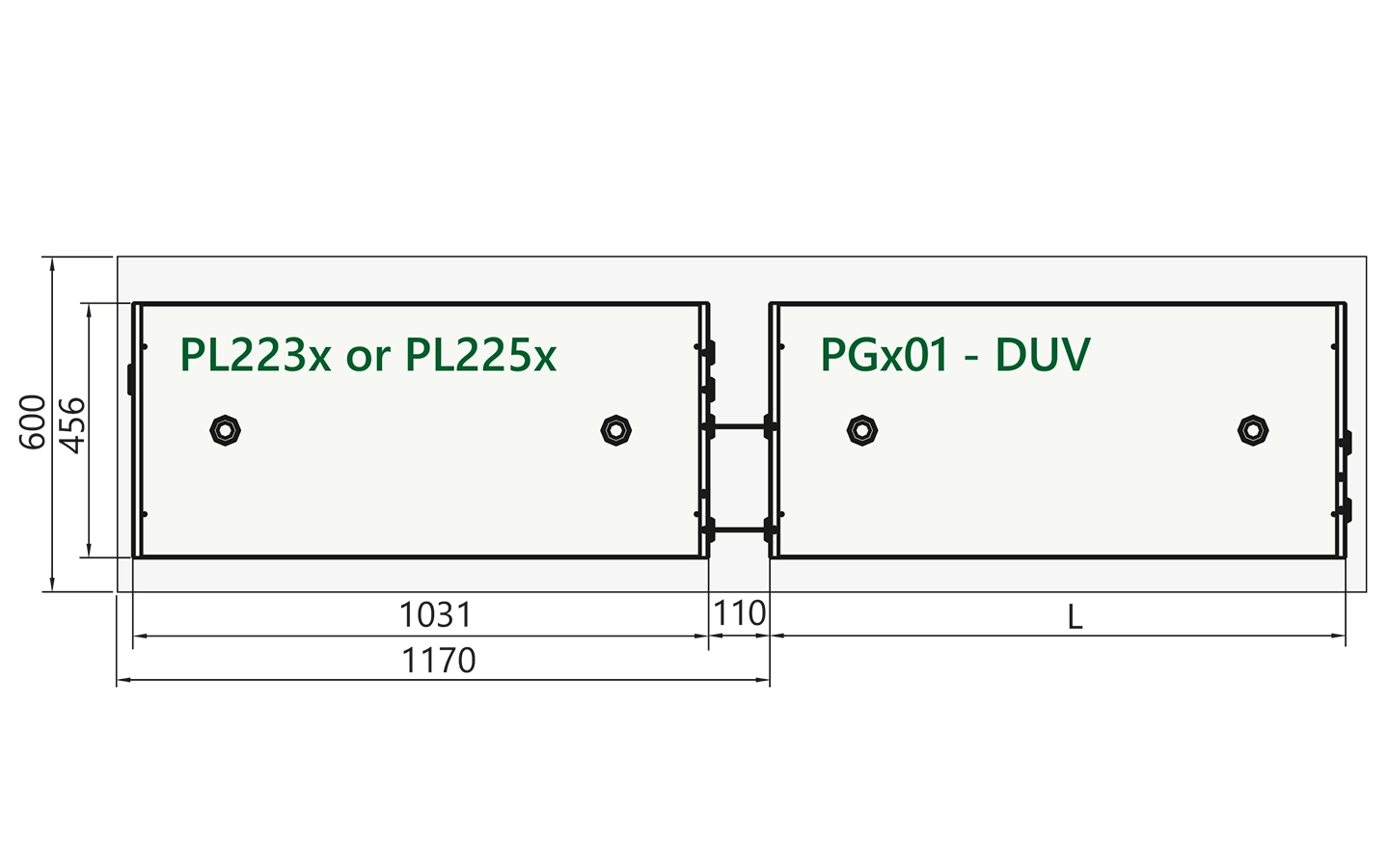
Arrangement of pump laser and PGx01-DUV unit on optical table.
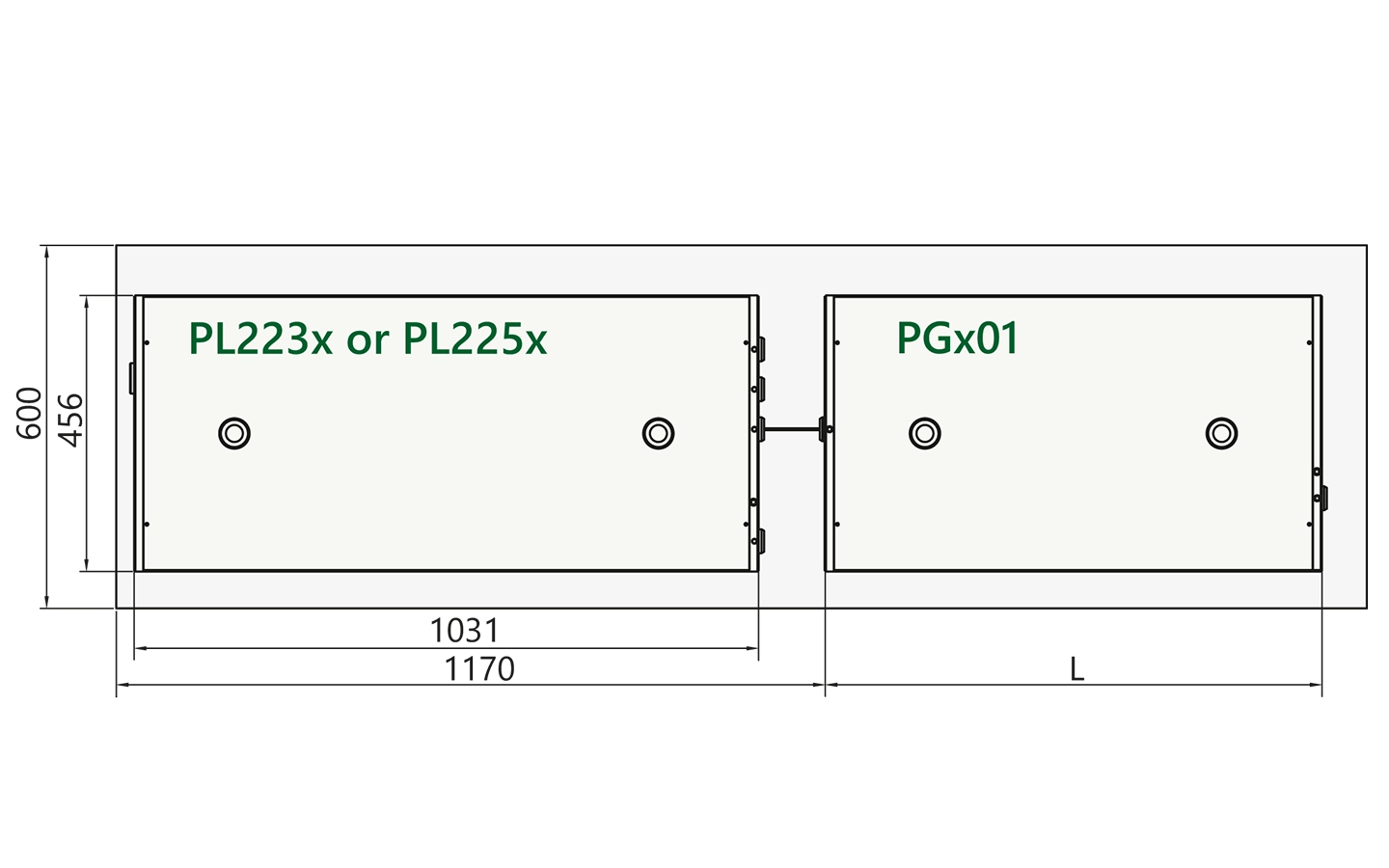
Arrangement of pump laser and PGx01 unit on optical table.