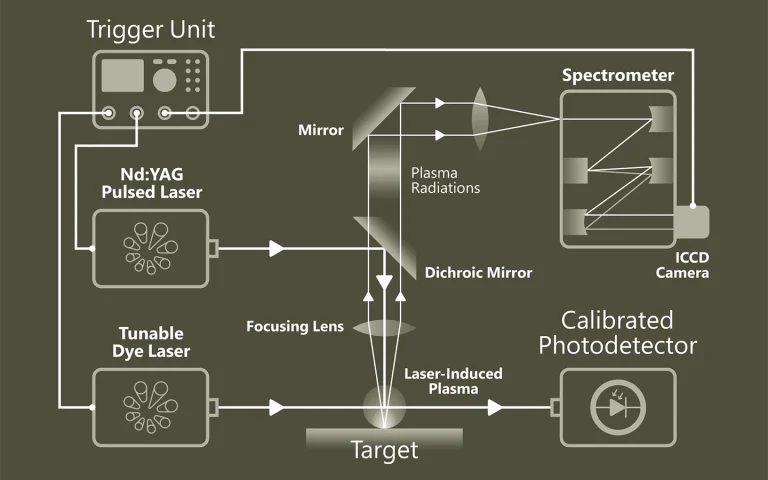
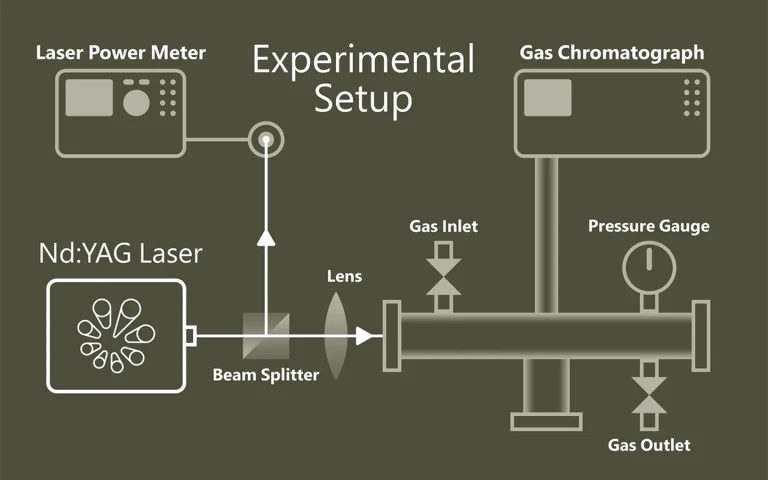
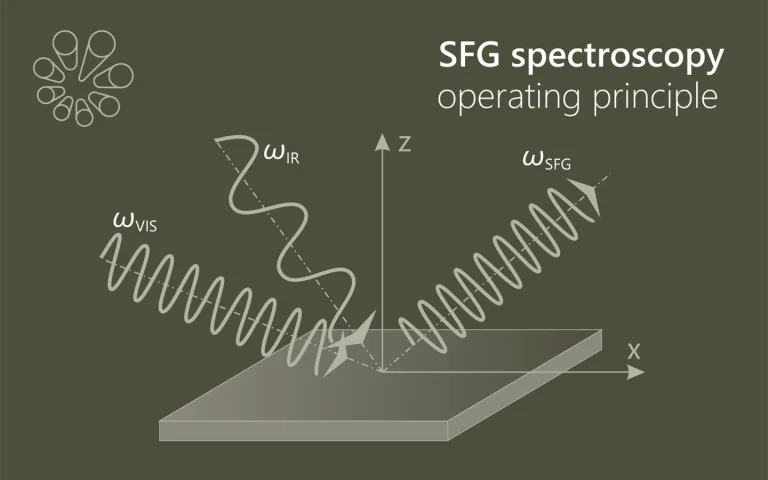
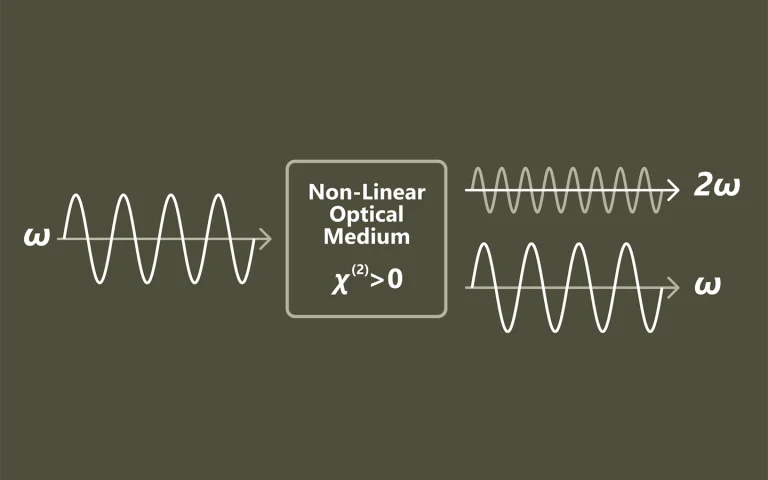
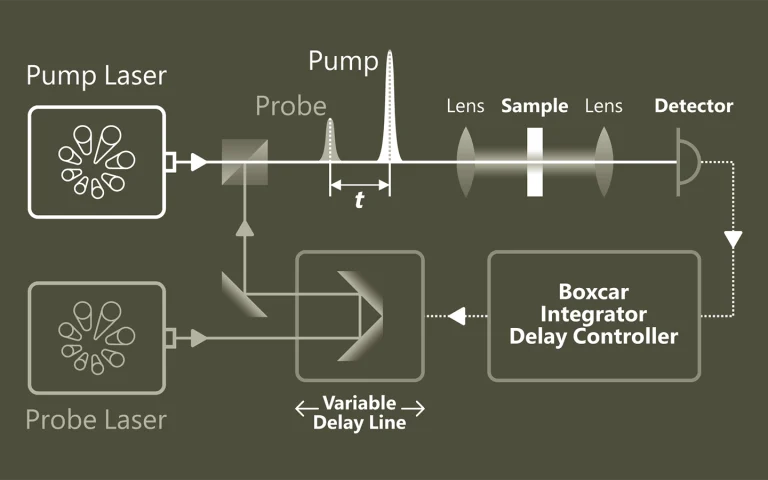
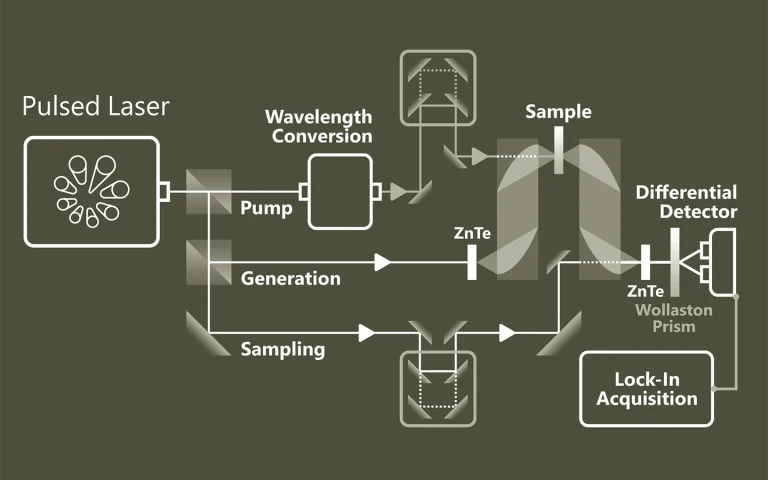
Pump-probe spectroscopy
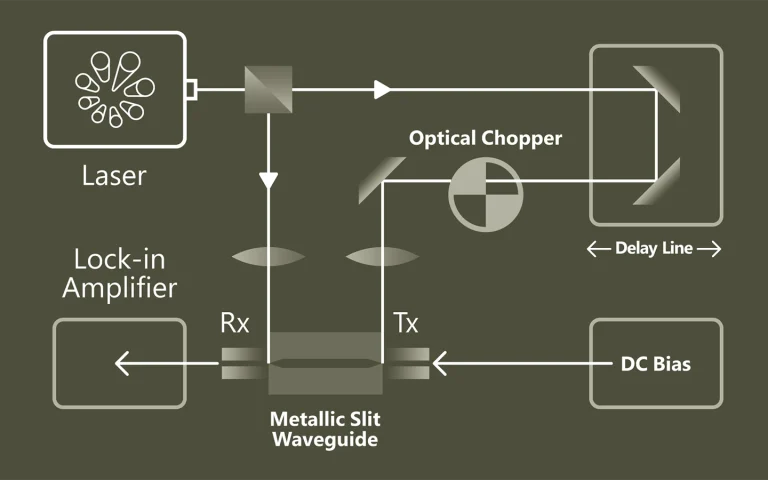
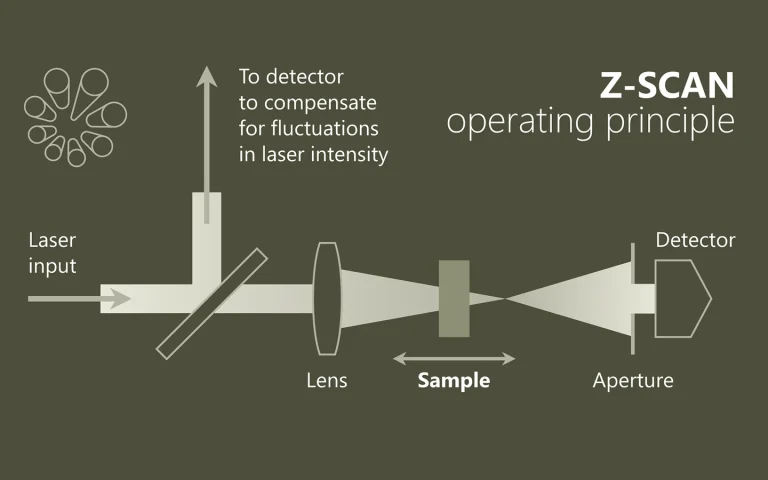
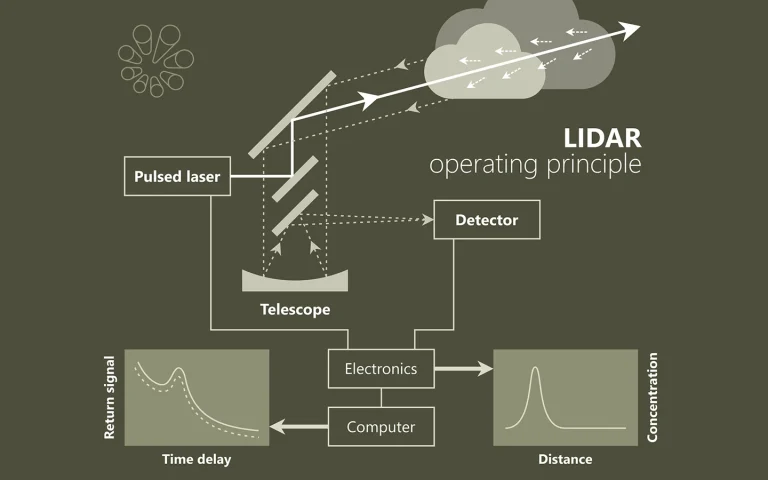
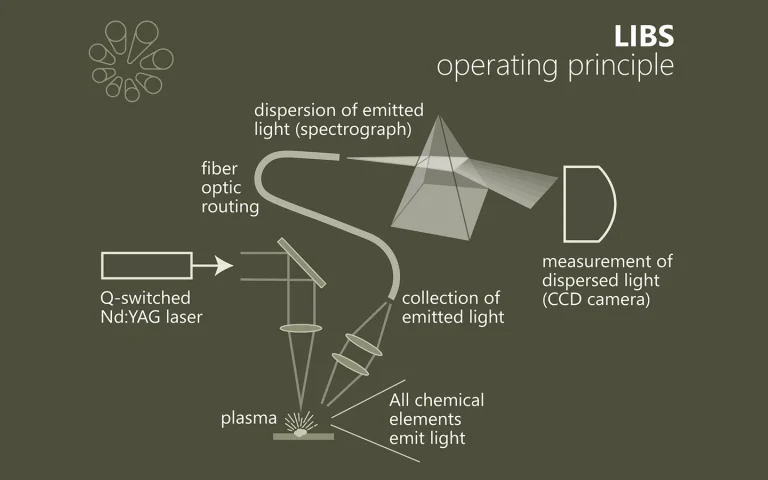
Ultrafast spectroscopy is based on the use of light pulses that have shorter temporal duration compared to the underlying dynamics of the system. Shorter pulse- better time resolution, longer pulse- better spectral resolution. Two pulses separated by time delay are used. First pulse excites (electronic and/or vibrational levels, ionises, make Coulombic explosions, creates plasma, etc.) sample, second delayed pulse probes- checks what happened at the certain time moment. Dynamics of investigated system is recorded by changing delay between pulses. Various experimental techniques are employed to record time-resolved signals: for example transient absorption, four-wave mixing, sum frequency generation. Pump and probe spectral range can vary from UV or even X-ray to far infrared.
The advantage of pump-probe spectroscopy is direct investigation of dynamics. For example: excitation relaxation, energy transfer, photochemical reactions dynamics and movement of particles, structural changes. Tunability of pump and probe pulses opens two dimensional pump-probe spectroscopy where is possible to obtain temporally resolved energy map of system which shows separated and coupled states.

Principle of Pump-Probe Spectroscopy.