High Energy Broadly Tunable Lasers NT340
High Energy Broadly Tunable Lasers
NT340 delivers hands‑free, no-gap tuning from from 192 nm to 4400 nm at up 20 Hz repetition rate from the one box. Featuring less than 5 cm‑1 linewdith and wide choice of options laser is excellent choice for very wide range of spectroscopic applications.

Features
- Customers recognized reliability
- Two years warranty
- Hands-free no gap wavelength tuning from 192 to 4400 nm *
- Up to 150 mJ pulse energy in visible spectral range
- Up to 22 mJ pulse energy in UV spectral range
- Up to 20 mJ pulse energy in MIR spectral range
- 3 – 5 ns pulse duration
- Up to 20 Hz pulse repetition rate
- Remote control via key pad or PC
- Optional separate shared output port for 532/1064 nm beam (separate output port for the 355 nm beam is standard)
- OPO pump energy monitoring
- Hermetically sealed oscillator cavity protects non-linear crystals from dust and humidity
* Automatic wavelength scan is programmable.
Applications
- Laser-induced fluorescence
- Flash photolysis
- Photobiology
- Remote sensing
- Time-resolved spectroscopy
- Non-linear spectroscopy
- Vibrational spectroscopy
- Cavity ring-down CRDS, cavity ring-down laser absorption CRLAS spectroscopy
- Infrared spectroscopy
- Gas spectroscopy
Benefits
- Hands-free wavelength tuning – no need for physical intervention
- The system is widely tunable
- 192 – 4400 nm and delivers high pulse energy (up to 150 mJ) that allows the investigation of an extensive range of materials
- Narrow linewidth (down to 3 cm‑1) and superior tuning resolution
- (1 – 2 cm‑1) allows recording of high quality spectra
- Flashlamps replacement without misalignment of the laser cavity saves on maintenance costs
- High integration level saves valuable space in the laboratory
- In-house design and manufacturing of complete systems, including pump lasers, guarantees on-time warranty and post warranty services and spares supply
- Variety of control interfaces: USB, RS232 and optional LAN, WLAN ensures easy control and integration with other equipment
- Attenuator and fiber coupling options facilitate incorporation of NT340 systems into various experimental environments
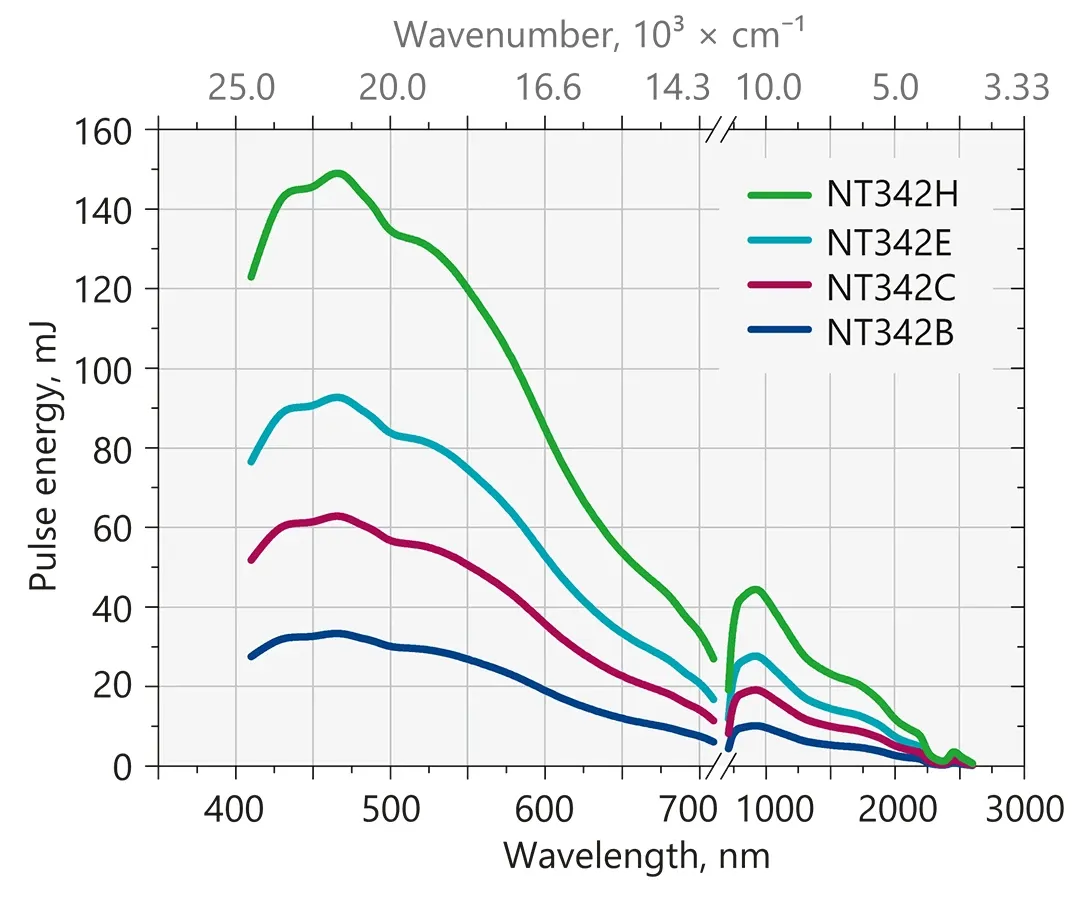
Typical output energy of the NT340 series tunable wavelength systems.
| Model | NT342B | NT342C | NT342E | NT342H |
|---|---|---|---|---|
| OPO specifications 1) | ||||
| Wavelength range 2) | ||||
| Signal | 410 – 710 nm 3) | 410 – 710 nm 3) | 410 – 710 nm 3) | 410 – 710 nm 3) |
| Idler | 710 – 2600 nm | 710 – 2600 nm | 710 – 2600 nm | 710 – 2600 nm |
| SH generator (optional) | 210 – 410 nm | 210 – 410 nm | 210 – 410 nm | 210 – 410 nm |
| SH/SF generator (optional) | 210 – 410 nm | 210 – 410 nm | 210 – 410 nm | 210 – 410 nm |
| DUV generator (optional) | 192 – 210 nm | 192 – 210 nm | 192 – 210 nm | 192 – 210 nm |
| MIR generator (optional) | n/a | 2500 – 4400 nm | n/a | n/a |
| Output pulse energy | ||||
| OPO 4) | 30 mJ | 60 mJ | 90 mJ | 150 mJ |
| SH generator (optional) 5) | 4 mJ | 6.5 mJ | 10 mJ | 15 mJ |
| SH/SF generator (optional) 6) | 6 mJ | 10 mJ | 15 mJ | 22 mJ |
| DUV generator (optional) 7) | 0.6 mJ | 1.2 mJ | 2 mJ | 3 mJ |
| MIR generator (optional) 8) | n/a | 20 mJ | n/a | n/a |
| Linewidth | < 5 cm‑1 9) | < 5 cm‑1 9) | < 5 cm‑1 9) | < 5 cm‑1 9) |
| Minimal tuning step 10) | ||||
| Signal (410 – 710 nm) | 1 cm‑1 | 1 cm‑1 | 1 cm‑1 | 1 cm‑1 |
| Idler (710 – 2600 nm) | 1 cm‑1 | 1 cm‑1 | 1 cm‑1 | 1 cm‑1 |
| SH/SF/DUV (192 – 410 nm) | 2 cm‑1 | 2 cm‑1 | 2 cm‑1 | 2 cm‑1 |
| MIR (2500 – 4400 nm) | n/a | 1 cm‑1 | n/a | n/a |
| Pulse duration 11) | 3 – 5 ns | 3 – 5 ns | 3 – 5 ns | 3 – 5 ns |
| Typical beam diameter 12) | 5 mm | 8 mm | 10 mm | 12 mm |
| Typical beam divergence 13) | < 2 mrad | < 2 mrad | < 2 mrad | < 2 mrad |
| Polarization | ||||
| Signal | horizontal | horizontal | horizontal | horizontal |
| Idler | vertical | vertical | vertical | vertical |
| SH/SF | horizontal | horizontal | horizontal | horizontal |
| DUV | vertical | vertical | vertical | vertical |
| MIR | n/a | horizontal | n/a | n/a |
| Pump laser 14) | ||||
| Pump wavelength | 355 nm | 355 nm | 355 nm | 355 nm |
| Typical pump pulse energy | 100 mJ | 150 mJ | 250 mJ | 400 mJ |
| Pulse duration | 4 – 7 ns | 4 – 7 ns | 4 – 7 ns | 4 – 7 ns |
| Beam quality | Hat-top in near field, without hot spots | Hat-top in near field, without hot spots | Hat-top in near field, without hot spots | Hat-top in near field, without hot spots |
| Beam divergence | < 0.6 mrad | < 0.6 mrad | < 0.6 mrad | < 0.6 mrad |
| Pulse energy stability (StdDev) | < 3.5 % | < 3.5 % | < 3.5 % | < 3.5 % |
| Pulse repetition rate | 10 or 20 Hz | 10 Hz | 10 Hz | 10 Hz |
| Physical characteristics | ||||
| Unit size (W × L × H) 15) | 456 × 821 × 270 mm | 456 × 821 × 270 mm | 456 × 821 × 270 mm | 456 × 821 × 270 mm |
| Power supply size (W × L × H) | 330 × 490 × 585 mm | 330 × 490 × 585 mm | 330 × 490 × 585 mm | 330 × 490 × 585 mm |
| Umbilical length | 2.5 m | 2.5 m | 2.5 m | 2.5 m |
| Operating requirements | ||||
| Water consumption (max 20 °C) 16) | < 10 l/min | < 10 l/min | < 10 l/min | < 10 l/min |
| Room temperature | 18 – 27 °C | 18 – 27 °C | 18 – 27 °C | 18 – 27 °C |
| Relative humidity | 20 – 80 % (non-condensing) | 20 – 80 % (non-condensing) | 20 – 80 % (non-condensing) | 20 – 80 % (non-condensing) |
| Power requirements | 200 – 240 VAC, single phase, 50/60 Hz | 200 – 240 VAC, single phase, 50/60 Hz | 200 – 240 VAC, single phase, 50/60 Hz | 200 – 240 VAC, single phase, 50/60 Hz |
| Power consumption | < 1.5 kVA (10 Hz) < 2.5 kVA (20 Hz) | < 1.5 kVA | < 1.5 kVA | < 2.5 kVA |
| Cleanliness of the room | not worse than ISO Class 9 | not worse than ISO Class 9 | not worse than ISO Class 9 | not worse than ISO Class 9 |
| Model | NT342B | NT342C | NT342E | NT342H |
|---|
- Due to continuous improvement, all specifications are subject to change. Parameters marked typical are illustrative; they are indications of typical performance and will vary with each unit we manufacture. Unless stated otherwise, all specifications are measured at 450 nm and for basic system without options.
- Hands-free tuning range is from 192 nm to 4400 nm. Up to 2500 nm idler tuning with MIR option.
- Tuning range extension to 400 – 709 nm is optional.
- Measured at 450 nm. See tuning curves for typical outputs at other wavelengths.
- Measured at 260 nm. See tuning curves for typical outputs at other wavelengths.
- Measured at 340 nm. SF generator is optimized for maximum output in 300 – 410 nm range. See tuning curves for typical outputs at other wavelengths.
- Measured at 200 nm. See tuning curves for typical outputs at other wavelengths.
- Measured at 2700 nm. See tuning curves for typical outputs at other wavelengths.
- Linewidth is <8 cm‑1 for 210 – 410 nm, 2500 – 4400 nm ranges.
- When wavelength is controlled from PC. When wavelength is controlled from keypad, tuning resolution is 0.1 nm for signal, 1 nm for idler, MIR and 0.05 nm for SH, SF and DUV.
- FWHM measured with photodiode featuring 1 ns rise time and 300 MHz bandwidth oscilloscope.
- Beam diameter is measured at 450 nm at the FWHM level. It is approximate and can vary depending on the pump pulse energy and wavelength.
- Full angle measured at the FWHM level at 450 nm, < 5 mrad at 3000 nm with MIR option.
- Separate output port for the 355 nm beam is standard. Outputs for 1064 nm and 532 nm beams are optional. Laser output will be optimized for the best OPO operation and specifications may vary with each unit we manufacture.
- Length from 821 to 1220 mm depending on configuration.
- Air cooled power supply is available as an option.
Note: Laser must be connected to the mains electricity all the time. If there will be no mains electricity for longer that 1 hour then laser (system) needs warm up for a few hours before switching on.
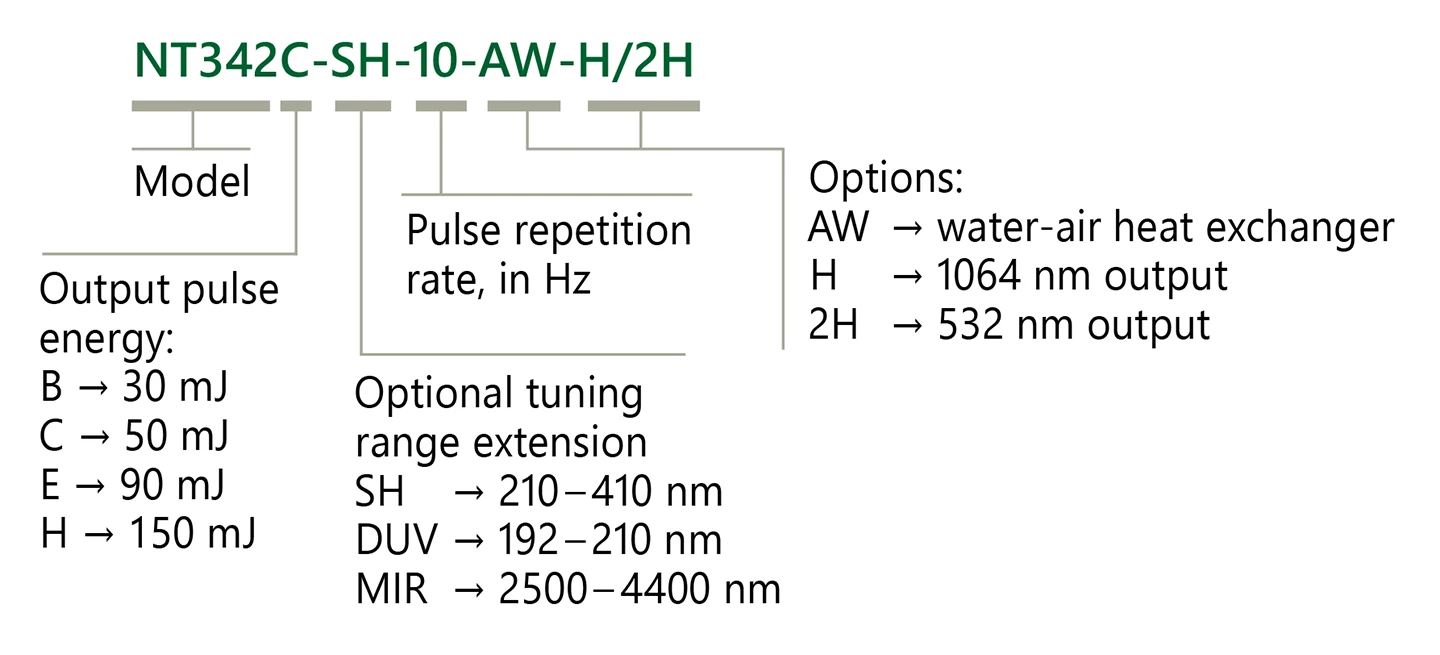
Ordering information of NT340 series lasers.
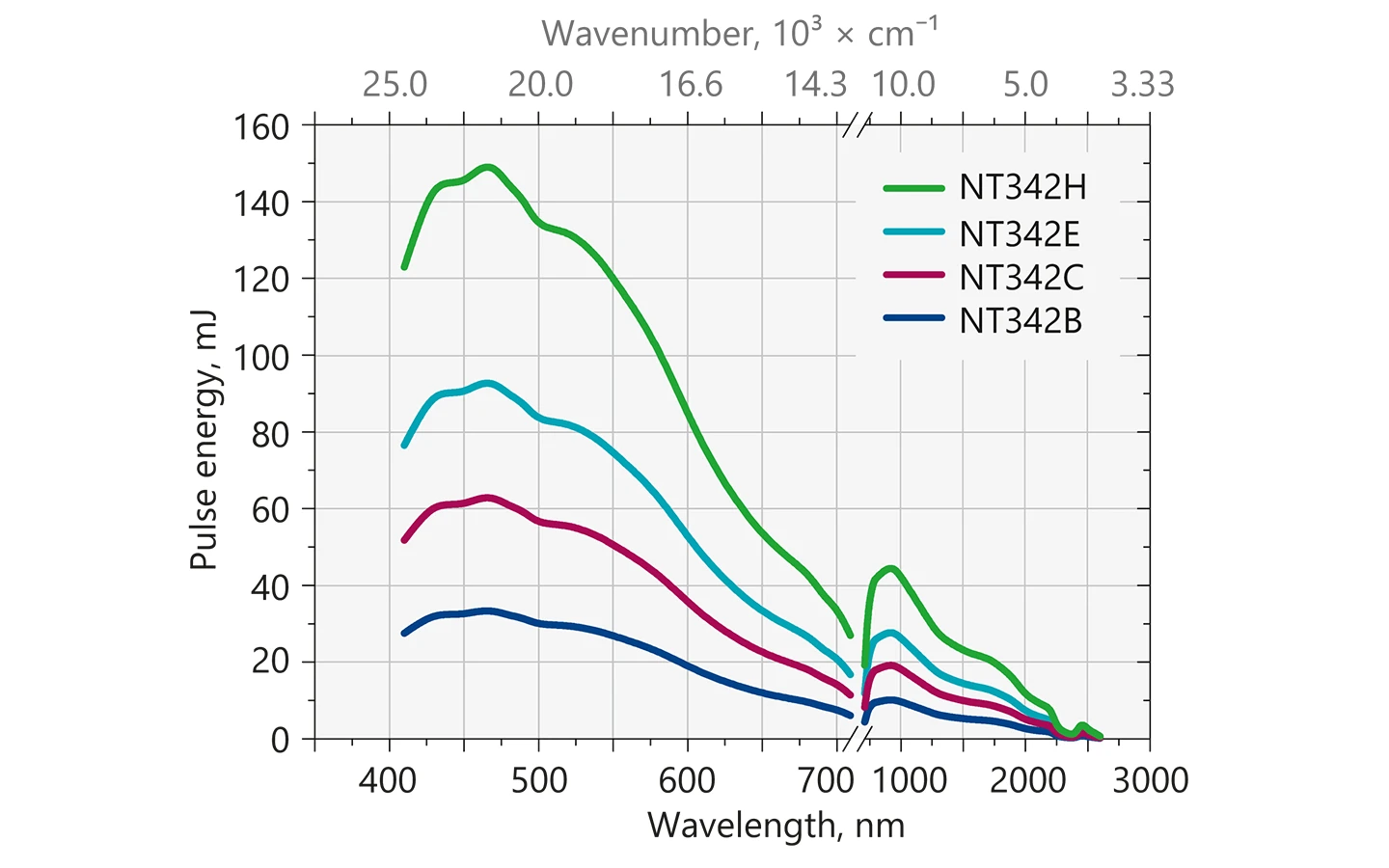
Typical output energy of the NT340 series tunable wavelength systems.
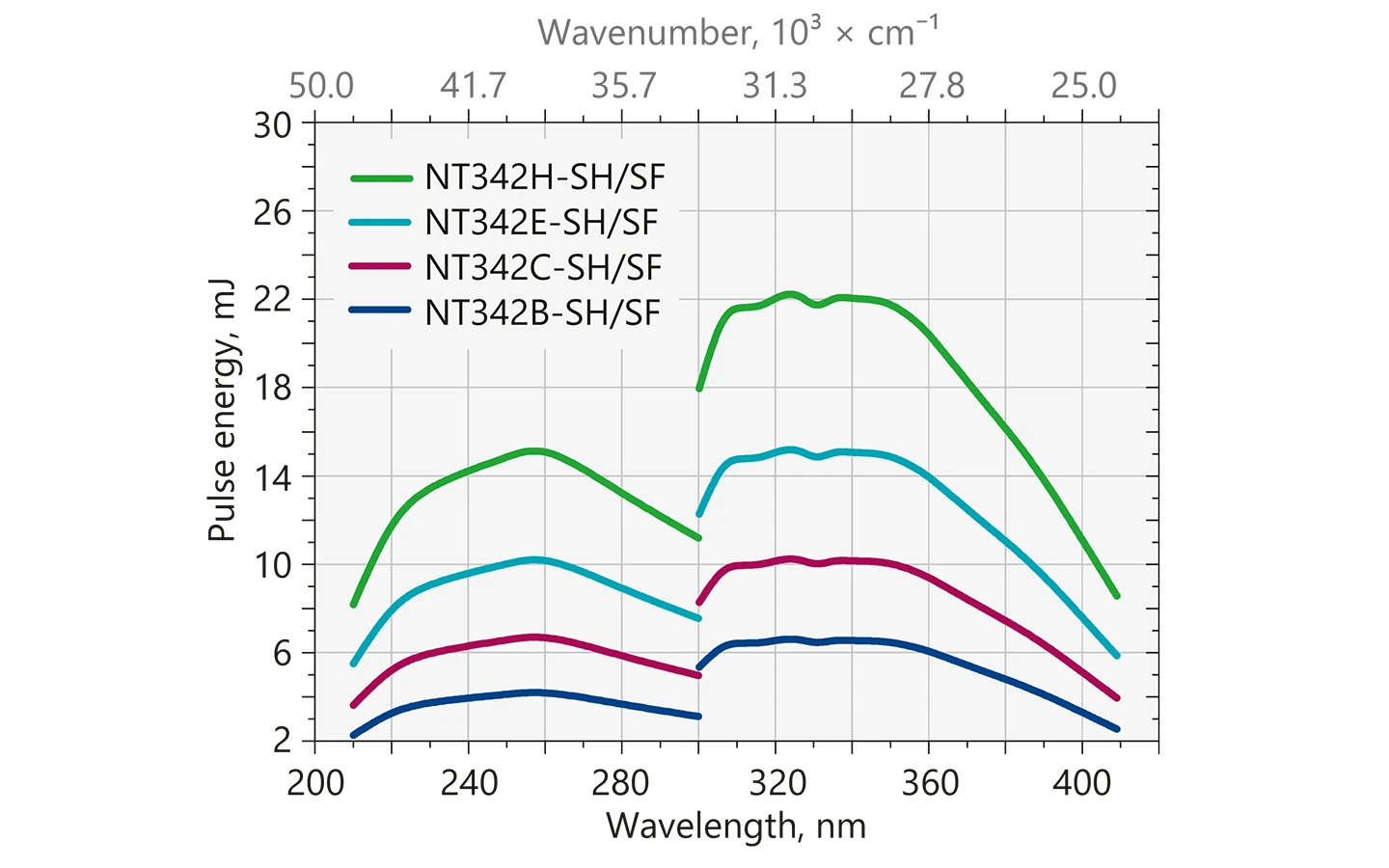
Typical output energy of the NT340 series tunable wavelength systems with SH/SF extension.
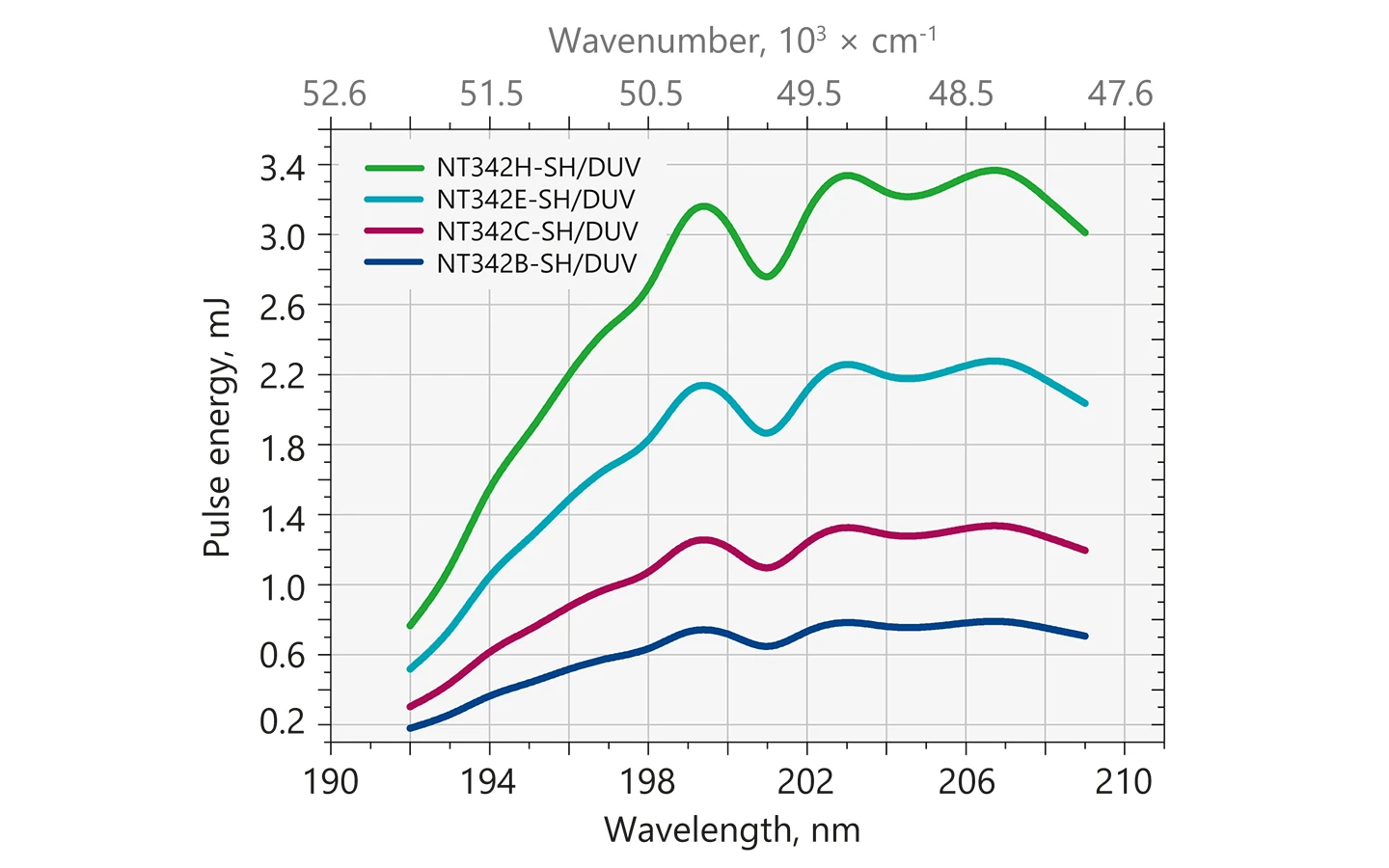
Typical output energy of the NT340 series tunable wavelength systems with SH/DUV extension.
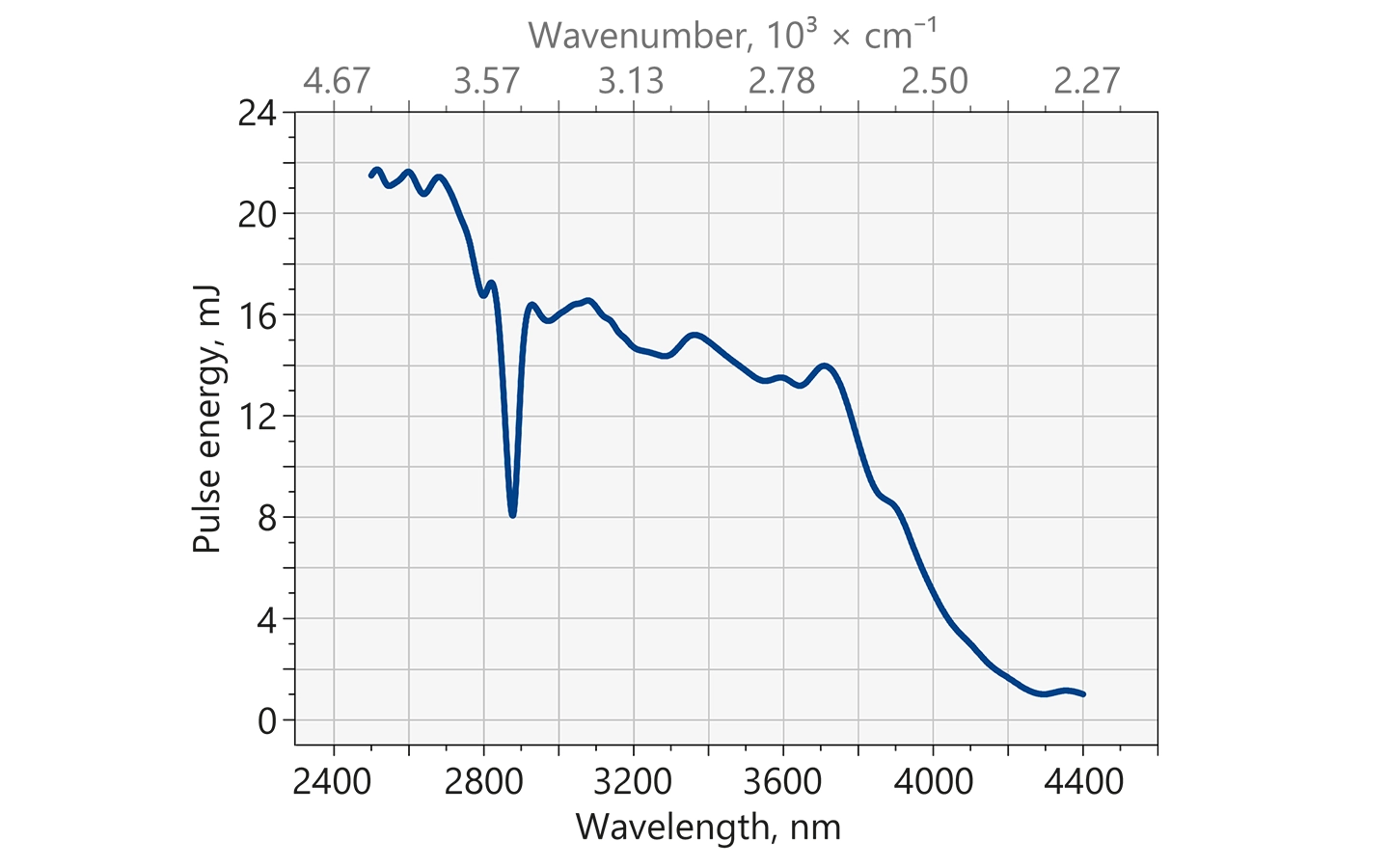
Typical output energy of the NT340 series tunable wavelength systems with MIR extension.

NT340 series laser typical beam profile at 450 nm after ~1.5 m distance from output.
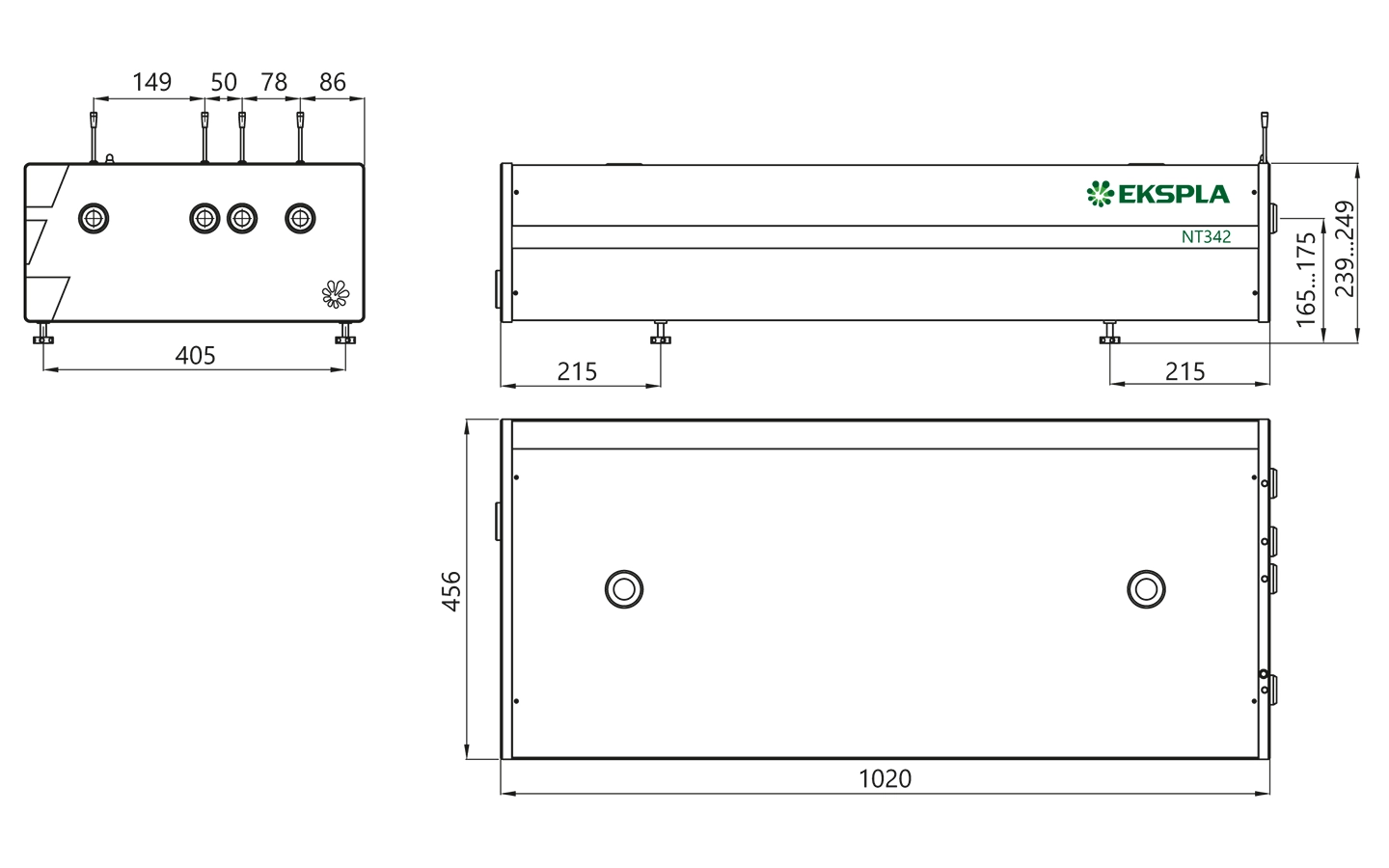
NT340 series laser head typical outline drawing.
Unit length and port position vary depending on model.
High energy broadly tunable laser NT342.